Medical Devices are any therapeutic or diagnostic agents or their accessories, which work on non-biochemical mode in the body. The primary scientific consideration in medical devices clinical trials is determination of ‘Which devices require clinical trial?” or “Does my device need clinical trial?” to undergo clinical studies.
WHICH MEDICAL DEVICES REQUIRE CLINICAL EVALUATION?
In Medical device life-cycle, preclinical bench-top analysis for mechanical and design tests; are the major determination criteria for acceptance of that device. It can be considered as Phase 0 studies for medical devices. Unlike the drugs, feasibility of medical devices practically is done in a combination of engineering tests and Animal studies. The pre Clinical studies become more important in medical device compared to drug because the evaluation of Medical devices (Both performance and safely) depends upon its adverse event occurrence (In contrast to drugs where Pharmaco-dynamics and tolerance are major safety endpoints and bioavailability and Improved life quality are performance outcomes).
From clinical research perspective, medical devices can be classified into four main categories-Therapeutic (Whether active or inactive), Non-therapeutic-non-diagnostic (Accessory), Diagnostic and Contraceptive medical devices.
The clinical trials for diagnostic devices are wider and diagnostic test outcome oriented. The clinical trials are required only to introduce new techniques. Same is the case with contraceptive devices. Contraceptive devices studies are mainly failure oriented single arm trials and accessories do not require clinical trial for regulatory or clinical evaluation.
The therapeutic medical devices, which mainly require clinical evaluation, have mainly two types, Implantable and external. The implantable devices includes Pacemaker, stents, arterio-venous fistulae, dental implants, orthopedic implants etc. and the non-implantable group include Cardio-Pulmonary Resuscitation (CPR) Devices , Heart and Lung Machine, Dialysis unit, Defibrillator (External) etc. Clinical trials in these Medical Devices are major concentrated on its performance and safety analysis. Most devices in this group need repeated confirmation of utility by clinical trials. Highly dynamic devices like stents hence require recurrent validation. Similar but relatively less severe is the case with a pace makers etc.
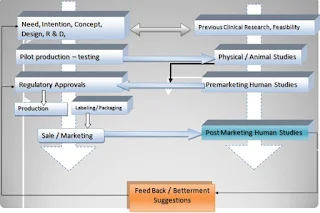 PHASES IN MEDICAL DEVICE CLINICAL TRIALS
PHASES IN MEDICAL DEVICE CLINICAL TRIALS
Unlike drug Clinical studies medical device clinical trials can be divided into three phases:
1. Pilot Clinical Trial
2. Pivotal Clinical Trials
3. Post Marketing servilence
|
Study types
|
Primary Intention
|
Desired outcomes
|
Points to be evaluated
|
|
First in human Studies
|
1. Feasibility of the device in Human subjects
|
1.Non-inferior performance over control arm
|
1. Logical Reasoning for determination of given endpoints (Cross
check animal data)
|
|
2. "No Additional Risk" profile of the device
|
2. No Additional risk over control arm
|
|
3. Performance of the device in target condition
|
|
|
|
4. The studies are performed in Simple population for
performance demonstration; have lowest risk
|
|
2. Other parameters if missed by study reporting (Source Animal
data)
|
|
Pivotal Studies
|
1. Safety and efficacy of the device in human subjects
|
"Safe for human wider human use"
|
1. Performance of the device in given end points (efficacy /
safety)
|
|
2. Performance of the device in target condition
|
2. Methodology of study
|
|
Post marketing
surveillance
|
Evaluation of Device
in more defined / Difficult subgroups
|
1. Performance and safety in More difficult conditions
|
1. Effect of parallel medication etc
|
|
2. Marketing tool
|
2. Study methodology
|
|
|
3. Preserved safety interest
|
CONSTRAINTS IN MEDICAL DEVICE CLINICAL TRIALS
Like drug, it is necessary to evaluate the safety and efficacy of medical device before for marketing approval. Apart from that medical devices will require performance evaluation, which makes the medical device studies more tricky compared to drug studies.
Another biggest constrain in most of the medical devices is lack of a perfect control. In most cases, earlier treatment by surgery, drug or previous generation device may also not prove a good control as indications in higher versions may differ from of the previous. Classical examples are randomization of stents against Bypass surgery, renal transplant against repetitive dialysis, Drug eluting liposuction versus pseudo-trimmer devices are all non-decisive. In most of these cases, even the design of the study was a challenging job. Even in previous versions of devices, there is similar propensity. DES versus metal stent, ERCP device versus laparoscopic cholecystectomy apparatus failed to achieve even primary endpoints due to lack of specificity in comparisons. Wise design of the study is important to mitigate such failure risks.
Apart from this, human safety is still a major clinical and ethical concern especially with implantable device related clinical procedures, as most of them are almost irreversible. Taking for example, a coronary stent, once implanted, even if it causes complications such as stent thrombosis, has no way to reverse the implant. Clinical studies for most of the therapeutic medical devices, unlike drugs, again are not possible in healthy subjects. Hence, there cannot be an “absolute safety” outcome for any medical device.
Another constraint is scope of error, which is very less in case of medical device clinical trial. Clinical data from all phases of medical devices is used as a very critical selection criterion by medical practitioners. Yet, making out-standing scientific value for the study and its outcome is difficult due to “lack of innovation” in many cases. Except a few clinical trials like REALTY, SYNTAX, LEADERS for DES, and a few more for Nephrological, neurological and urological devices, there are too few innovative designs. Achieving “Superiority” is also difficult task in medical device trial. Pertaining to all these major points, there is a lot of opportunity for young scientists to invest their mind in making several developments.
Indian Clinical research industry is progressively developing and adding newer segments to its overall expansion. In last few years, Medical Devices have made a significant share in the overall statistics. Considering the regulatory scenario, most of these trials take place simply on a notification. A few CROs like Genelife in Mumbai, have taken initiatives in making medical device based clinical trials more innovative and standard. Even if in current time there are no many pilot/pivotal studies and FIM studies taking place, we see closest future having wider prospects and scopes for Medical devices clinical research in India.
Dhirendra V. Singh
Managing Director
GENELIFE CLINICAL RESEARCH
33,34,37 Ground Floor, Cinewonder Mall, Ghodbandar Road, Thane West-400607. INDIA
Telephone: +91-(0)22-65242666 begin_of_the_skype_highlighting +91-(0)22-65242666 end_of_the_skype_highlighting Mob: +91-(0)9819055580 begin_of_the_skype_highlighting +91-(0)9819055580 end_of_the_skype_highlighting Fax: +91 22 25841316
Email: info@genelifecr.com Website: http://www.genelifecr.com/, Blog:http://genelifecr.blogspot.com/














